'아두카누맙' 이어 두 번째 치매 신약
에자이 '약값 연간 약 3400만원 책정'
피플바이오·젬백스 등 관련주 상승세
다만 임상 3상 중 사망 사고 지켜봐야

일본 제약사 에자이가 선보인 치매 신약 레카네맙이 미국 식품의약국(FDA)으로부터 가속 승인을 받으면서 국내 관련주가 들썩이고 있다. 하지만 일각에선 임상 시험 기간 발생한 사망사고 등 부작용 문제를 신중하게 지켜봐야 한다는 우려 섞인 목소리도 나온다.
9일 여성경제신문 취재를 종합해보면 최근 레카네맙은 FDA로부터 신약 허가를 받았다. 레카네맙의 상표명은 레켐비로 확정됐다. 이로써 레카네맙은 지난해 아두카누맙에 이어 두 번째 알츠하이머 치료제가 됐다.
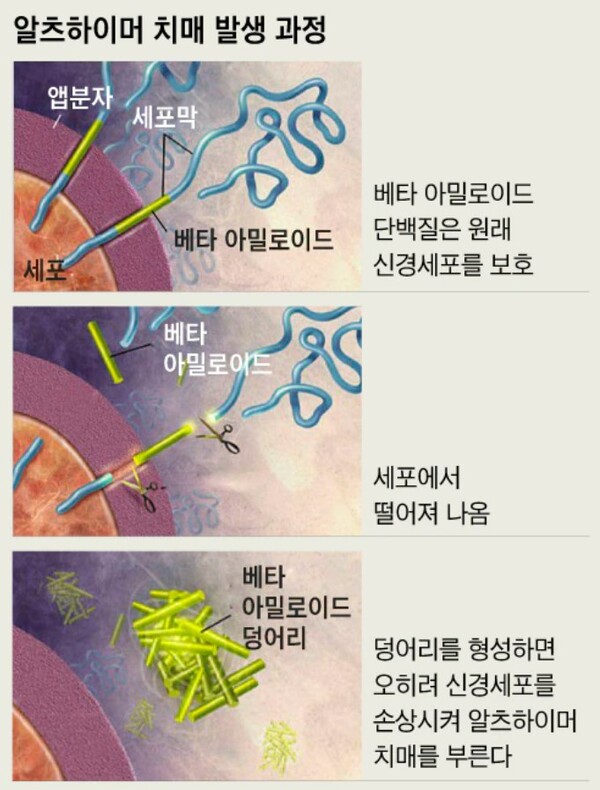
이번 승인으로 초기 알츠하이머성 치매 환자는 리카네맙을 사용할 수 있게 됐다. 에자이에 따르면 해당 신약은 치매 발병 원인으로 알려진 뇌 속 '아밀로이드 베타'라고 불리는 단백질을 뇌에서 제거하는 것을 표적으로 삼는다. 치매 발병 원인은 현재까지도 확실하게 알려진 바 없지만, 아밀로이드 베타 단백질이 쌓이는 것이 가장 큰 원인으로 지목되고 있다.
레카네맙은 임상 3상에서 인지력 저하 증상을 약 27% 줄인 것으로 나타났다. 또한 레카네맙 약값은 연간 2만 6500달러(약 3400만원) 수준으로 책정됐다고 에자이 측은 밝혔다. 미국 FDA 승인에 따라 에자이는 중국과 유럽, 일본 등에서도 품목 허가를 추진한다는 입장이다.
이 같은 호재 속에 피플바이오와 메디프, 삼진제약, 젬백스 등 국내 치매 치료제 관련주도 덩달아 상승세다. 피플바이오 주가는 레카네맙의 FDA 가속 승인이 결정된 시점인 한국시간 6일 오전 9시 30분 기준 1만4400원에서 9일 오전 10시 30분 기준 1만8650원 선까지 올랐다.
같은 기간 메디프론 주가도 1135원에서 1625원, 삼진제약은 2만4350원에서 2만5050원, 젬백스 또한 1만1200원에서 1만1850원으로 상승 폭을 키웠다. 국내 바이오주에 대한 영향력에도 관심이 쏠린다. 레카네맙이 출시되면 삼성바이오로직스와 같은 기업이 치료제 위탁생산 업체로 선정될 가능성이 있는 만큼 국내 바이오 시장에 미치는 영향도 부정할 수 없기 때문이다.
증권가 전망도 밝다. 박재경 하나증권 연구원은 "이번 신약 소식에 따른 제약·바이오 섹터 전반의 멀티플을 기대하기보다 위탁생산 등 연구·개발(R&D) 성과를 기대할 수 있는 일부 기업에 대한 투자 전략이 유효하다"라고 말했다.
아두카누맙 실패 딛고 올라선 레카네맙··· 부작용 이슈 괜찮을까
레카네맙 승인에 앞서 에자이와 바이오젠은 세계 최초 치매 신약으로 아두카누맙을 지난해 선보였다. 다만 약효 논란과 FDA 심사 과정에서의 부적절한 유착 관계 의혹 등이 제기되면서 시장에서 아두카누맙은 외면받고 있다.
특히 레카네맙의 경우 임상 시험 단계에서 사망자가 발생하면서 안전성 논란도 불거졌다. 최근 사이언스지에 따르면 레카네맙 임상 3상에서 세 번째 환자 사망이 보고된 바 있다. 이에 대해 네덜란드 라이덴 대학 신경학과 반 에텐 교수는 사이언스지와의 인터뷰에서 "환자는 뇌의 부종, 출혈 등으로 인해 사망했다"며 "(레카네맙) 약물의 심각한 부작용일 수 있다"고 봤다.
매튜 슈라그 미국 밴더빌트 대학 신경과학과 교수도 "에자이와 바이오젠이 이 사건을 공개하지 않은 것은 보고된 안전성 데이터가 완전하다는 확신을 훼손한다"고 말했다. 에자이가 지난달 주요 알츠하이머 학회에서 레카네맙의 3상 시험 데이터를 설명하면서 사망자 관련 정보를 자세히 공개하지 않았다는 것이다.
에자이 측은 임상 연구에 참여하는 환자의 개인 정보 보호 약속에 따라 구체적인 사망 정보를 공개할 수 없다는 입장이다. 지난해 11월 알츠하이머 학회에서 임상 중 사망과 관련 내용을 공개하지 않은 것은 현재까지 제공된 정보로는 사망과 관련이 있다는 의심을 하기 어렵고, 확장 연구의 데이터 분석이 완료되지 않았기 때문이라고 에자이는 설명했다. 에자이 측은 레카네맙 임상 시험 중 발생한 첫 번째와 두 번째 환자의 사망 원인도 약물과 관련이 없다고 밝힌 바 있다.
바이오젠과 에자이는 지난달 미국 샌프란시스코에서 열린 '2022 알츠하이머 임상학회(CTAD)'에서 레카네맙 임상 3상 시험 결과를 공개했다.
이에 따르면 알츠하이머 환자 1795명을 투약그룹과 위약그룹으로 나눈 뒤 2주에 한 번씩 레카네맙과 위약을 각각 투여한 결과, 18개월 후 투약 군의 68%에서 아밀로이드 베타 단백질이 제거됐다. 레카네맙을 투여한 환자들의 임상치매척도(CDR-SB) 개선 정도 역시 위약군에 비해 27% 높았다.
유효성 측면에서는 유의미한 결과를 얻었지만, 안전성 측면에서는 여전히 논란이 있었다. 레카네맙 투여군의 21.3%가 뇌부종 등 아밀로이드 관련 영상 이상(ARIA)을 호소했으며, 위약군에서 발생 비율은 9.3%였다.
아두카누맙의 경우 투여군의 41%에서 ARIA가 보고돼 심각한 우려가 제기돼 유럽의약품청(EMA)으로부터 승인을 거부당했다. 레카네맙은 이보다는 낮으나 여전히 높은 ARIA 발생률이 나타난 것으로 평가됐다. 다만 바이오젠 측은 레카네맙의 ARIA 발생률이 예상 범위 이내였다고 설명했다.
국내 치매 학계에선 레카네맙에 대한 장기간 연구가 더 필요하다고 봤다. 대한치매학회 관계자는 여성경제신문과 통화에서 "레카네맙이 초기 알츠하이머병 환자에게 효과적이고 아두카누맙보다 이상 반응이 적은 것으로 나타났다"며 "레카네맙 만큼 효과를 보인 약제가 없다는 점에서 FDA 정식 허가를 받게 된 것"이라고 전했다.
다만 "연구 추적관찰 기간은 18개월로 레카네맙군의 치료기간이 길어질수록 위약군과의 효과 차이가 벌어졌다"면서도 "레카네맙 효과가 언제까지 유지될지 그리고 부작용과 관련한 정확한 데이터는 2년 이상 추적관찰이 필요하며, 현재 이를 확인하려는 오픈라벨 연장연구가 진행 중"이라고 덧붙였다.